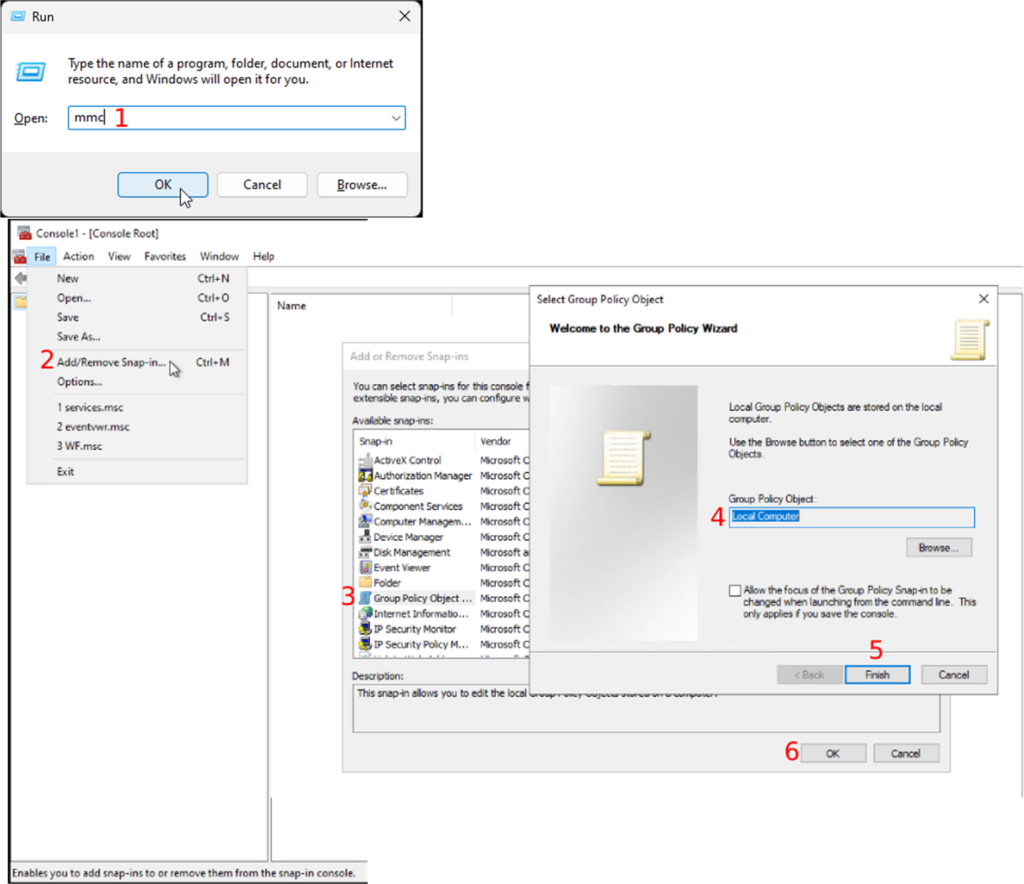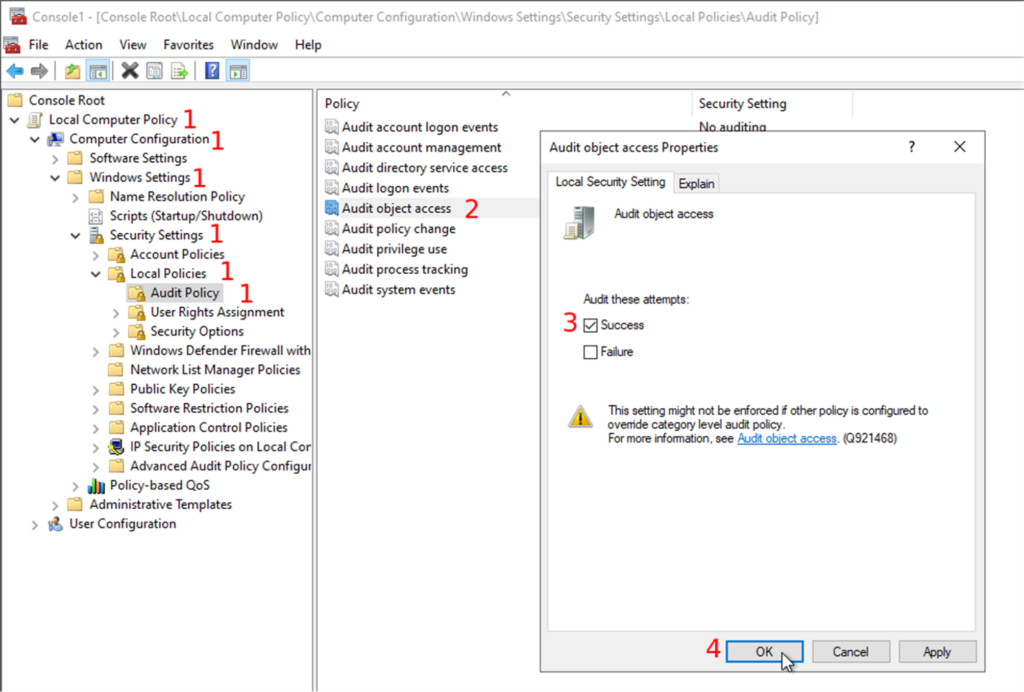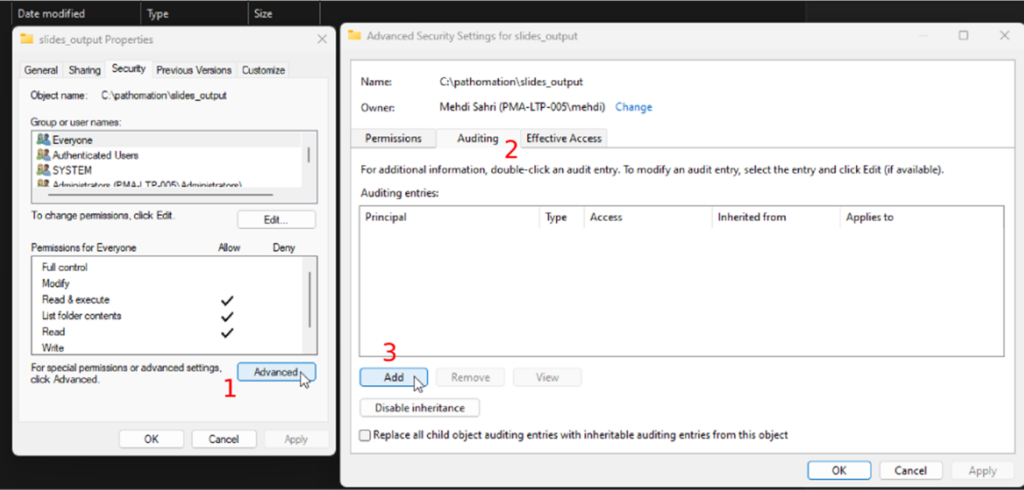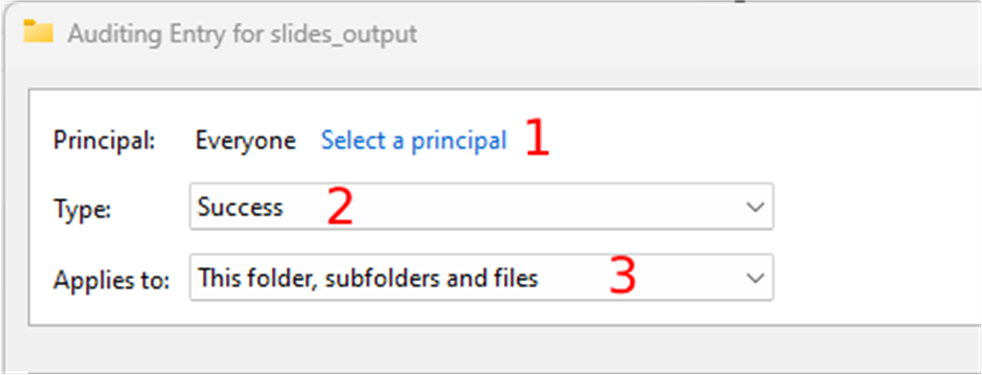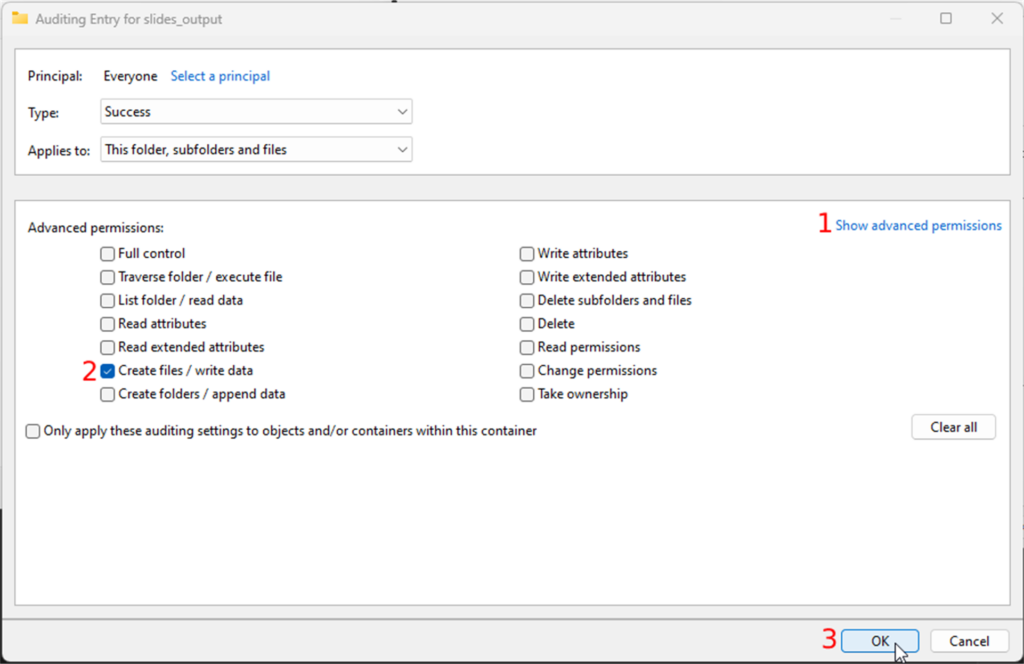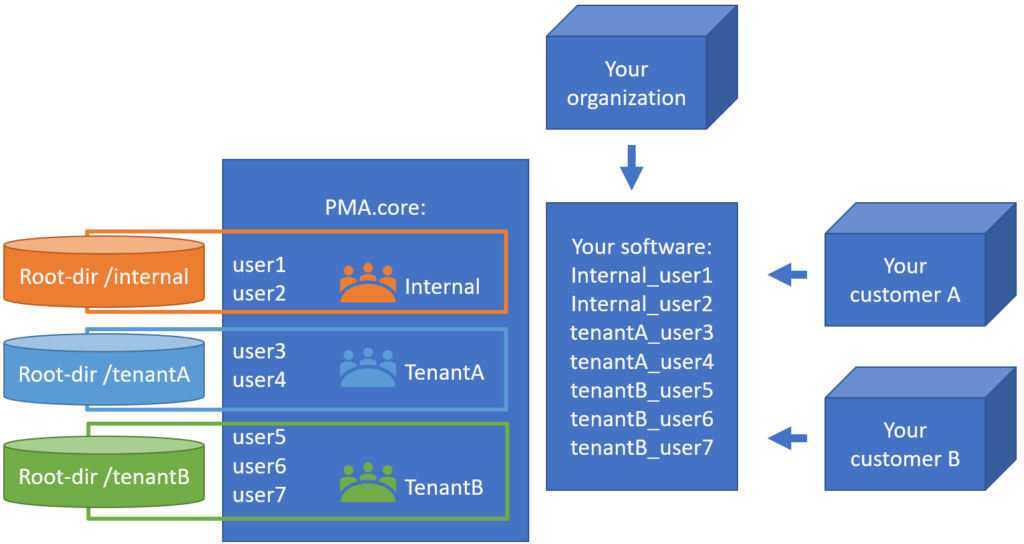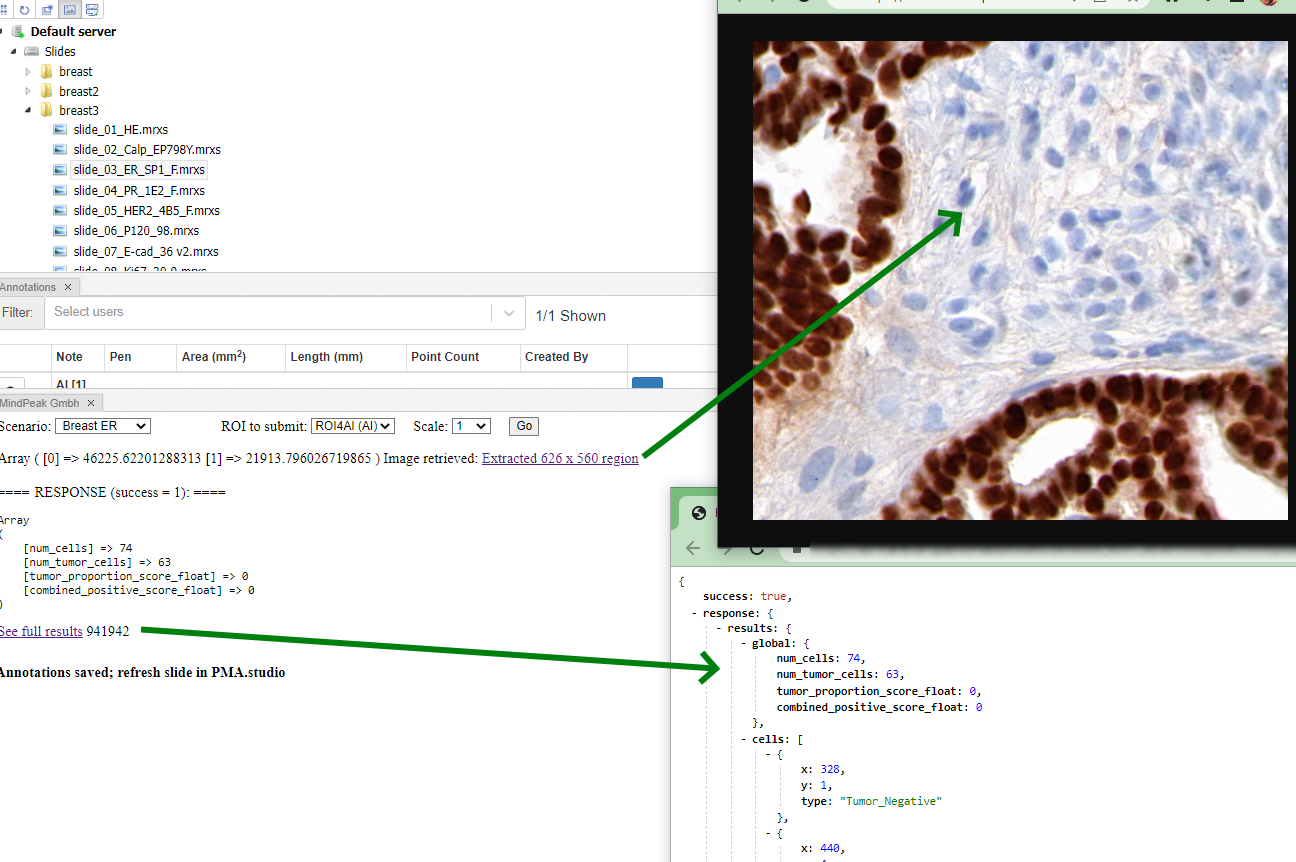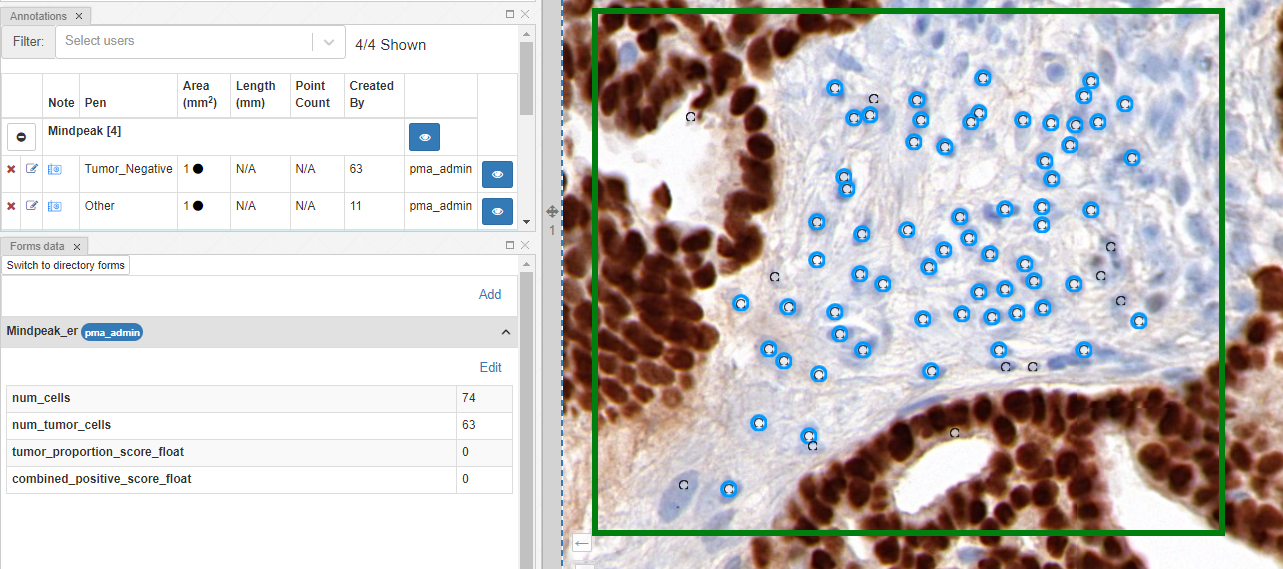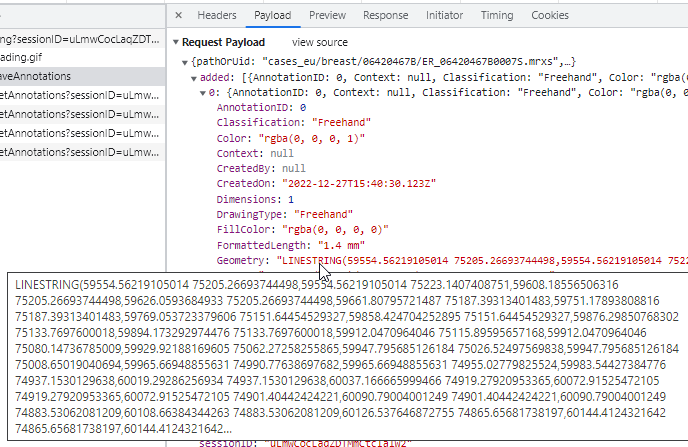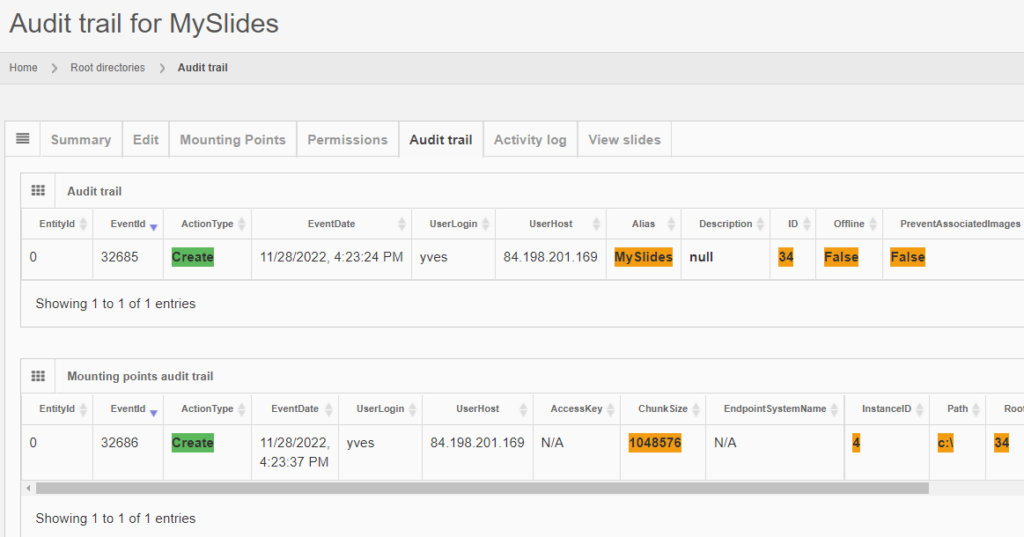
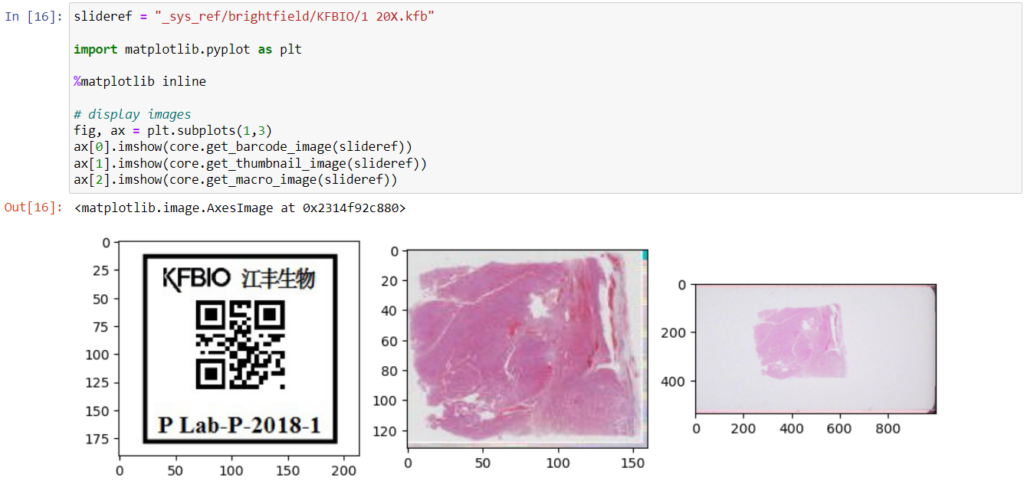
Whole Slide Images not only contain pyramidical giga-pixel images, but also various lower-resolution images with helpful information.
Image metadata
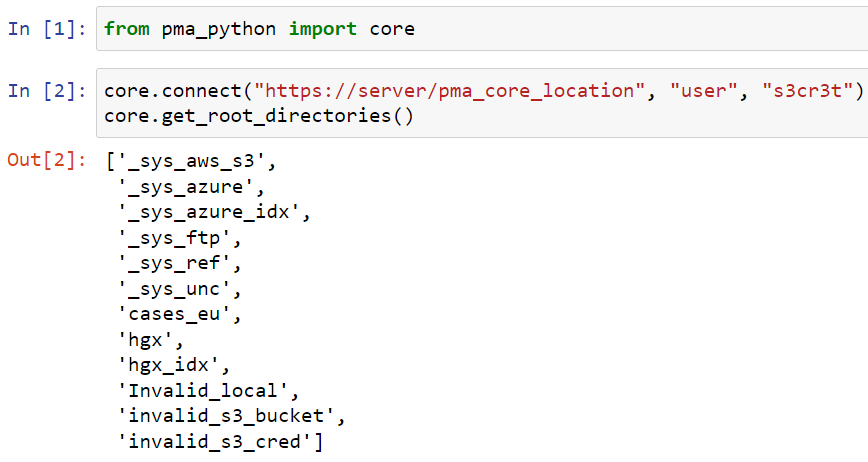
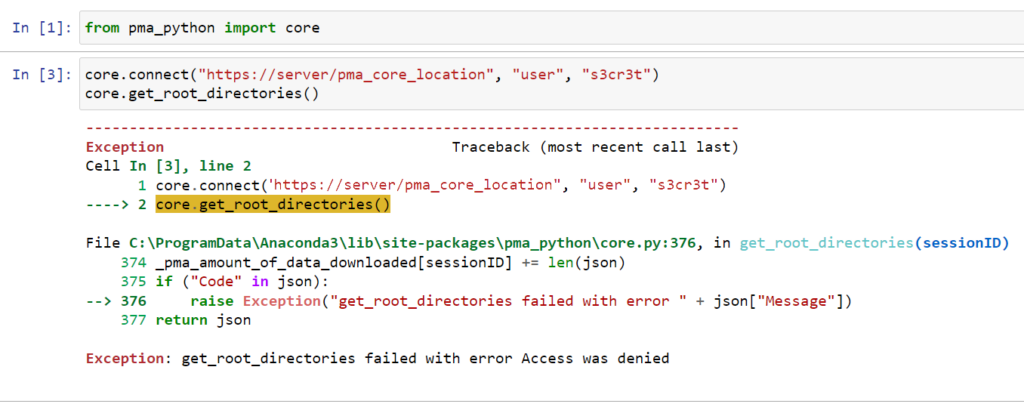
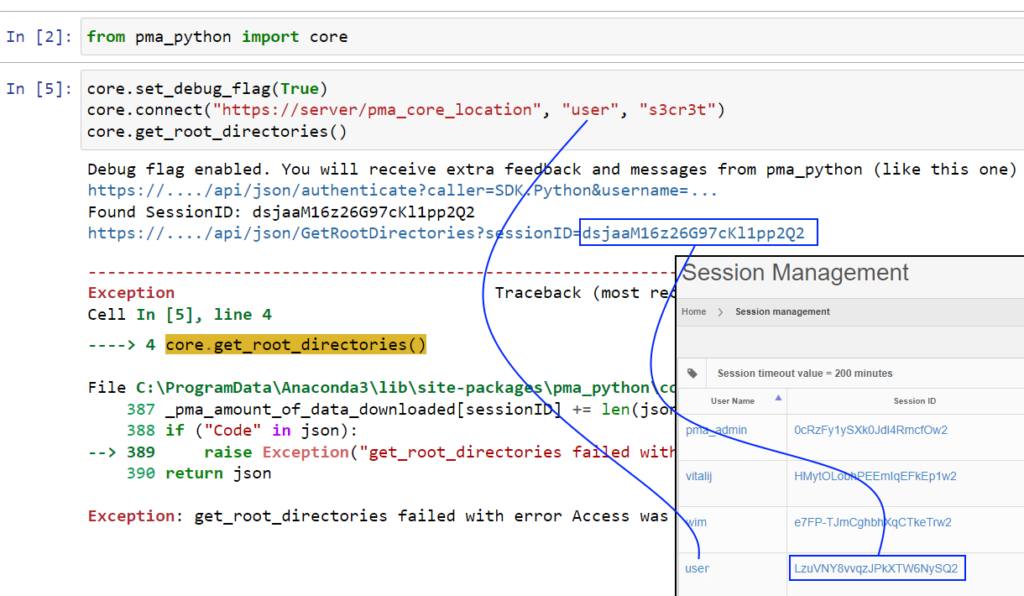
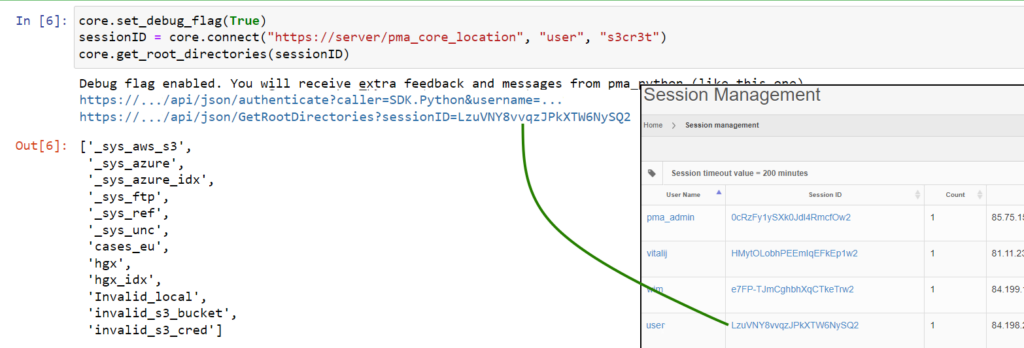
The type of image data that can be retrieved from an individual virtual slide is described at https://docs.pathomation.com/pma.core/3.0.2/doku.php?id=nonapi.
Three of these endpoints can be classified as image metadata:
- Thumbnail: A thumbnail image of the scanned slide. A low-resolution representation of the area of the glass slide that was scanned.
- Macro: A macro image of the entire slide (typically generated by a separate low-resolution camera that’s part of the slide scanner)
- Barcode: the label pasted on the slide. It’s possible for this paper label to contain a barcode. If this is the case, it is possible to interpret the barcode through a separate API call.
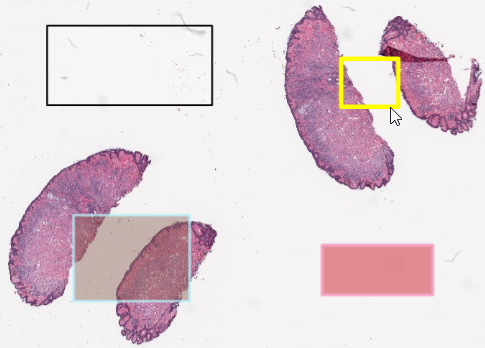
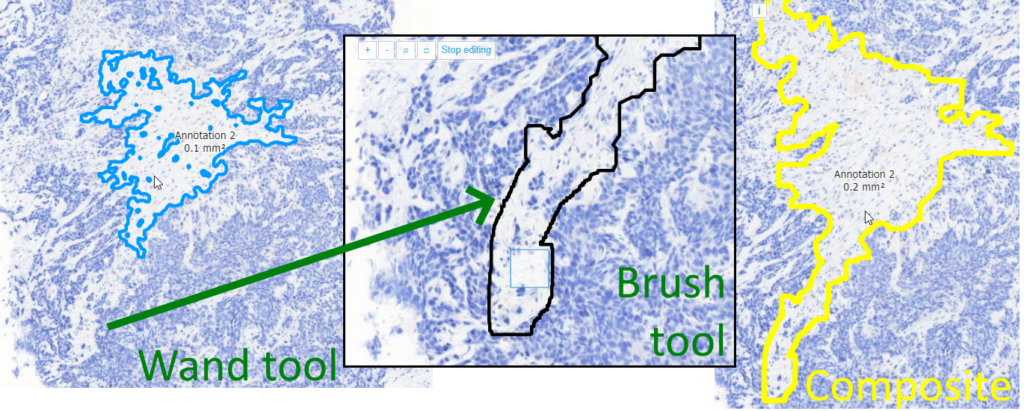
Here’s what all three images can look like for a particular slide:
The tile and region endpoints are related to selective pixel extraction and are irrelevant for the context of this article.
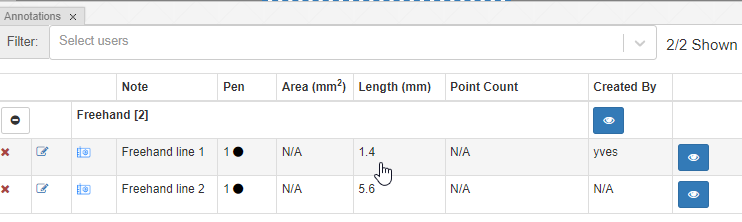
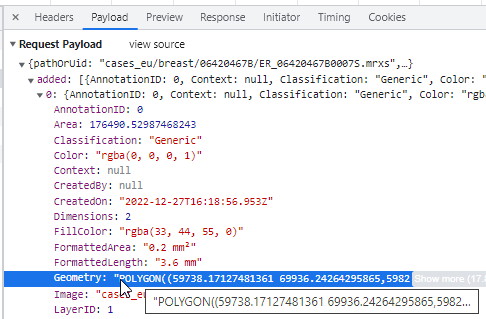
Macro images
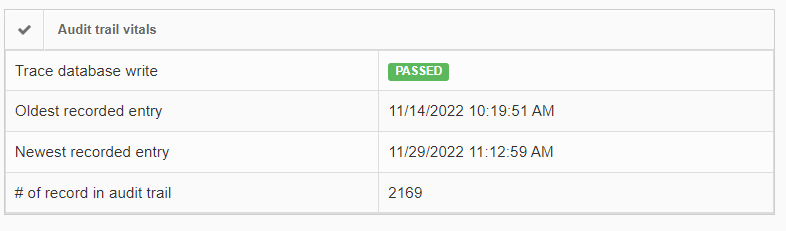
The macro image shows the entire glass slide and offers an overview of all the tissue fragments present on the slide. A thumbnail represents the area of the slide scanned at high resolution. In case the tissue recognition of the scanner missed a tissue fragment, the tissue visible in the thumbnail will not match with the macro. Checking for mismatches in tissue between macros and thumbnails is an important quality control step in digital pathology.
But whole slide image are complex data structures. Not all file formats from all vendors therefore support all possible features and not all file formats from all vendors contain all metadata described here. This is obvious for the JPEG and PNG file formats.
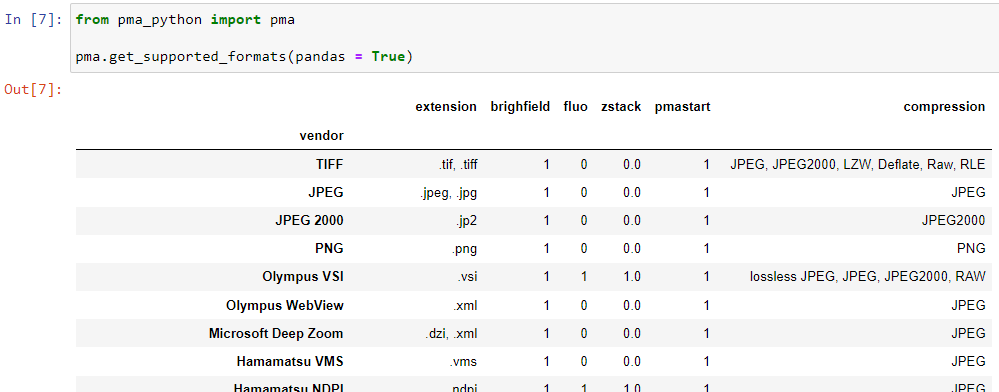
Currently PMA.core supports macro images for twelve file formats:
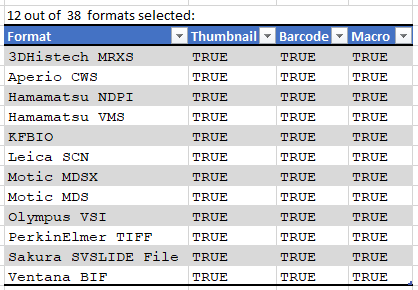
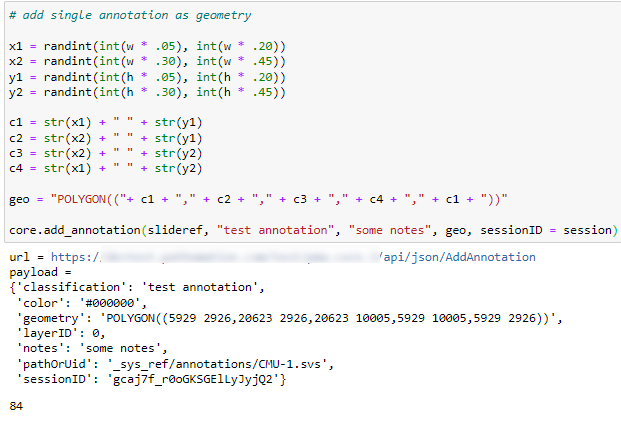
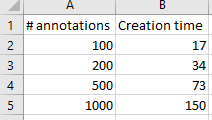
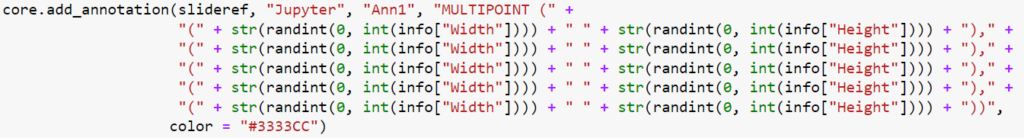
And here’s the code that we used to determine that output:
import pandas as pd
from pma_python import core
core.connect("http://server/pma.core/", "us3rn4m3", "S3CR3T!")
format_features = {}
def get_associated_image_types(slide):
info = core.get_slide_info(slide)
return info["AssociatedImageTypes"]
def get_file_format(slide):
info = core.get_slide_info(slide)
for item in info["MetaData"]:
if (item["Name"].lower() == "fileformat"):
return item["Value"]
return ""
slides = core.get_slides("_sys_ref/brightfield", recursive = True)
for slide in slides:
fmt = get_file_format(slide)
if len(fmt) > 0:
if not(fmt in format_features):
format_features[fmt] = {"Thumbnail": False, "Barcode": False, "Macro": False}
for tp in get_associated_image_types(slide):
format_features[fmt][tp] = True
formats = pd.DataFrame.from_dict(format_features, orient="index")
macro_formats = formats[formats["Macro"] == True]
print(len(macro_formats), "out of ", len(formats), " formats selected:")
print(macro_formats)
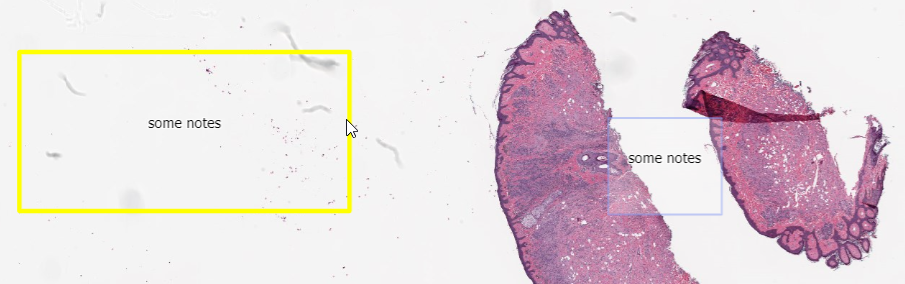
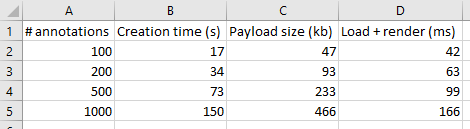
We can write PHP code that generates a catalog of all slide features in a single overview:
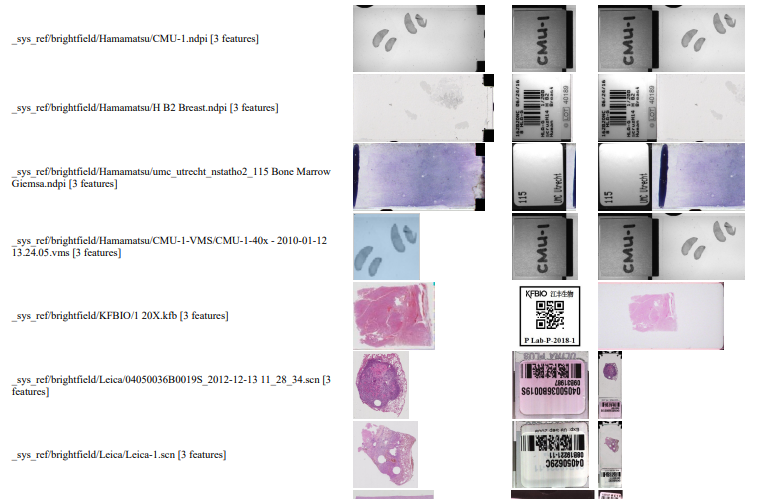
Note that the orientation of the macro-images can vary from one file format to the next: some opt for a vertical positioning, others for a vertical one. All of this can be compensated for, but it has repercussions: for example, if you rotate the macro images from Leica, it is useful for rotate the thumbnails the same way; otherwise comparison between macro and thumbnail becomes complicated.
Also note that with some vendors, the macro image is interpreted as “complete slide surface, including barcode”, whereas for others the macro image is the glass area of the slide, minus the label area.
The PHP source code reads like this:
<html>
<head><title>All meta features</title></head>
<body>
<?php
require_once "php/lib_pathomation.php";
Use Pathomation\PmaPhp\Core;
$session = Core::connect("https://server/pma.core/", "us3rn4m3", "S3CR3T");
// echo "PMA.core SessionID = ".$session."<br>";
?>
<table>
<tr><th>Format</th><th>Barcode</th><th>Thumbnail</th><th>Macro</th></tr>
<?php
$slides = Core::getSlides("_sys_ref/brightfield", $session, true
foreach ($slides as $slide) {
$info = Core::getSlideInfo($slide, $session);
$fc = count($info["AssociatedImageTypes"]);
if ($fc > 2) {
echo "<tr>
<td>$slide [".$fc." features]</td>
<td><img src='".Core::getBarcodeUrl($slide, $session)."' height=100></td>
<td><img src='".Core::getThumbnailUrl($slide, $session)."' height=100></td>
<td><img src='".Core::getMacroUrl($slide, $session)."' height=100></td>
</tr>";
}
}
?>
</table>
</body>
</html>
Image encoding hack (how to generate comprehensive reports)
You could use the above script as a starting point to generate daily overview reports of what’s been scanned. The output is “just” HTML in a browser, but comes with a disclaimer: Depending on the method used to convert the live page into an actual static report, may loose the images in the process.
A way around this is to switch from URL reference to base64 image encoding; like this:
function includeThumbnail($slide, $session) {
$img = Core::getThumbnailImage($slide, $session);
$img_data = base64_encode($img);
return "data:image/jpg;base64,".$img_data;
}
The result of using this code is that the HTML file is now a single self-contained document. You can use it as an attachment, or share it through instant messaging (IM), without the need for anybody to have access to underlying image files or access to the original PMA.core tile server.
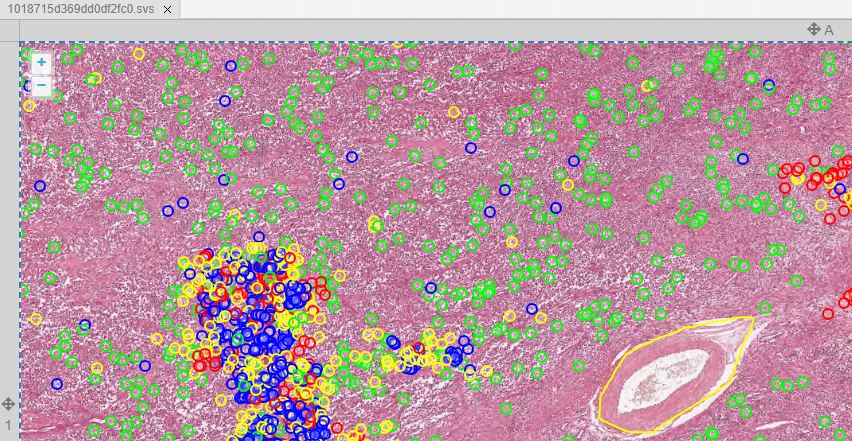
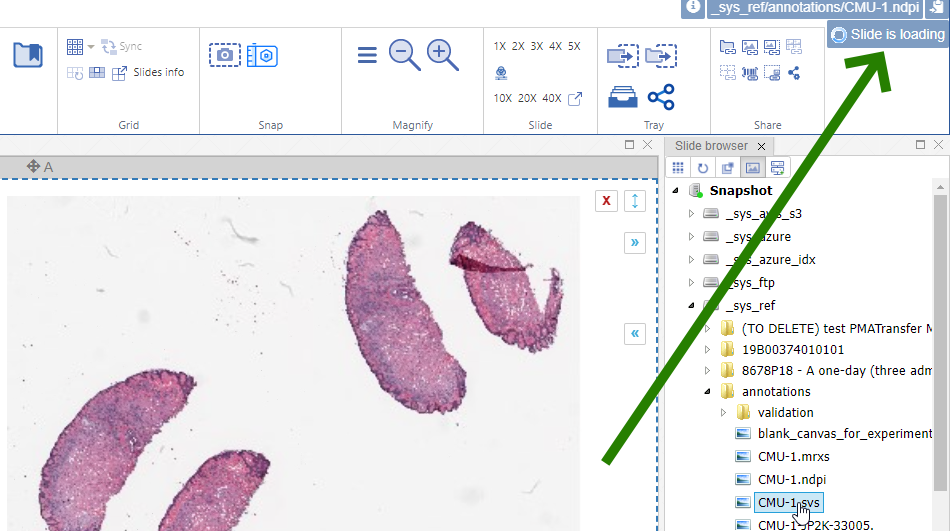
Interactive demo
We have an interactive demo available, too. This one was built with the JavaScript PMA.UI framework:
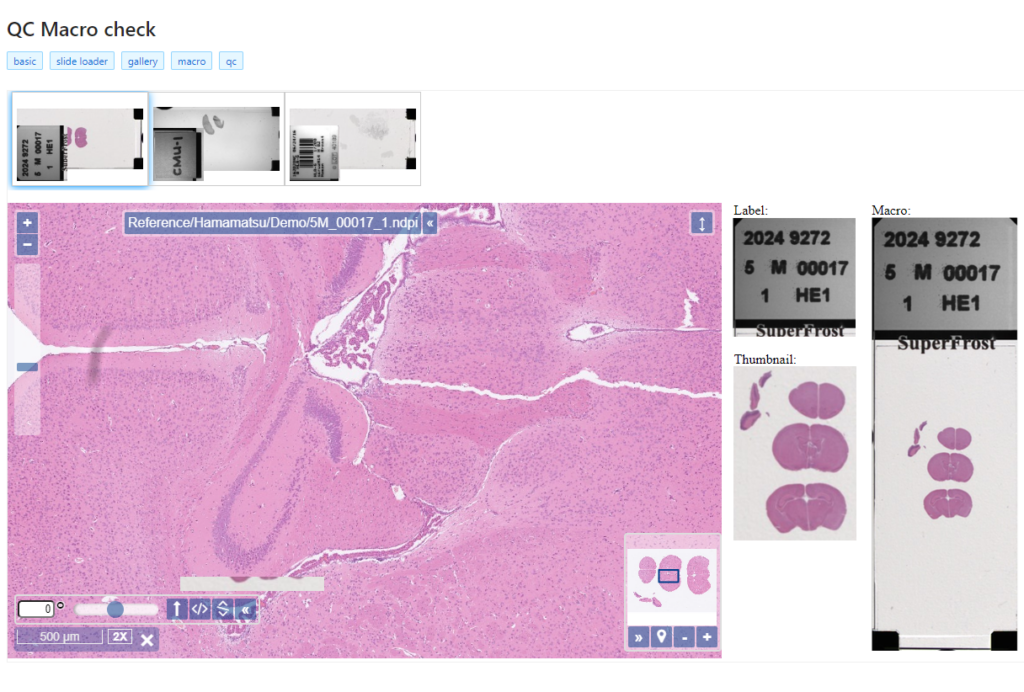
The demo is available through our JavaScript demo portal. We offer it as an inspiration as to what a custom QC module could look like.
In terms of workflow automation, you could write an algorithm that does tissue detection on both the thumbnail image, as well as the macro image, and compares the results. If the result is too divergent (or if the area of detected tissue falls below a pre-determined threshold), you could flag the slides as possibly faulty and send them to a lab technician for closer inspection.
Platform meta-data
Our SDKs are designed in such a way that they encapsulate typical plumbing code that would otherwise be needed to invoke the API-calls directly. When we do our job right, your Python script shouldn’t need the requests library anymore; your Java application shouldn’t need the java.net namespace anymore.
But our Pathomation platform itself has meta-data, too: like the file formats it supports.
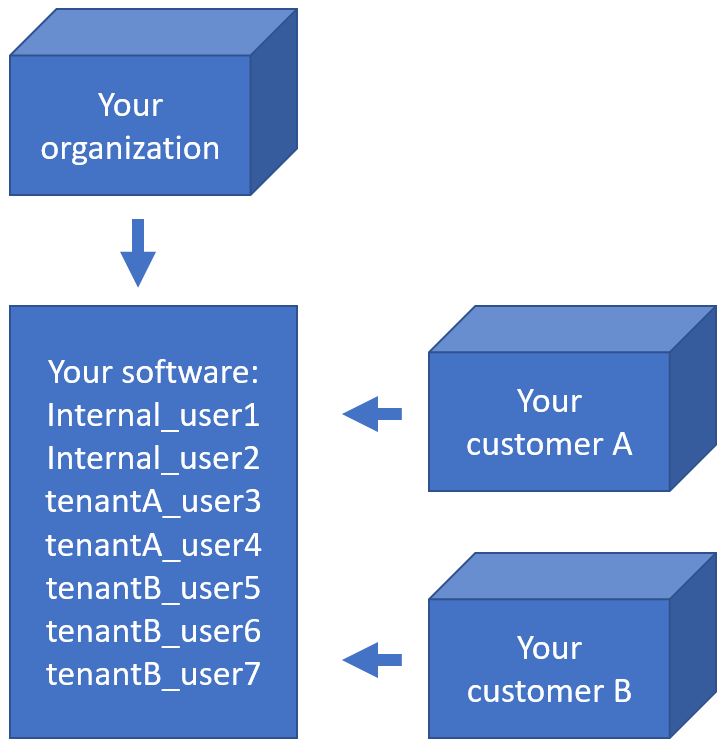
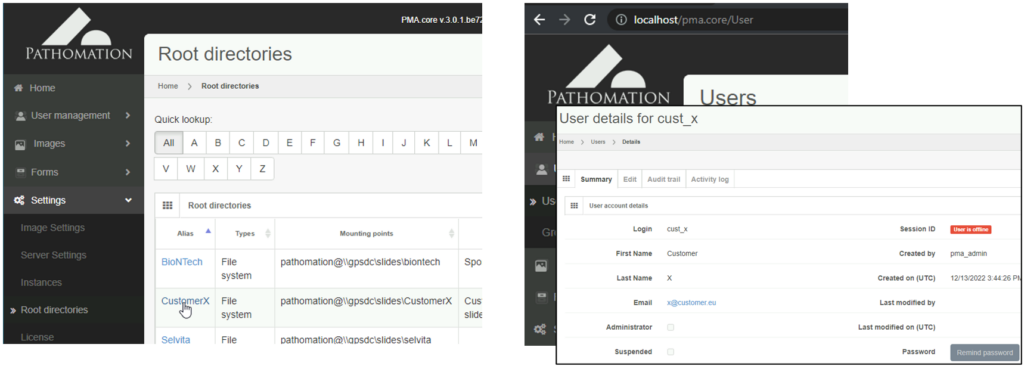
When you’re an OEM solution provider that’s relying on our SDK to incorporate digital pathology features in your own software, you may want to offer a way for your own customers to see what file formats you support (through PMA.core then).
Therefore, we recently added a new call so people can programmatically request what modalities of what file formats we support. You get a JSON structure back, and you can render it to your own liking, which results in a nicer and smoother user experience on your end. We’re using this ourselves in PMA.transfer (as an alternative to opening a new webbrowser and pointing to https://www.pathomation.com/formats):
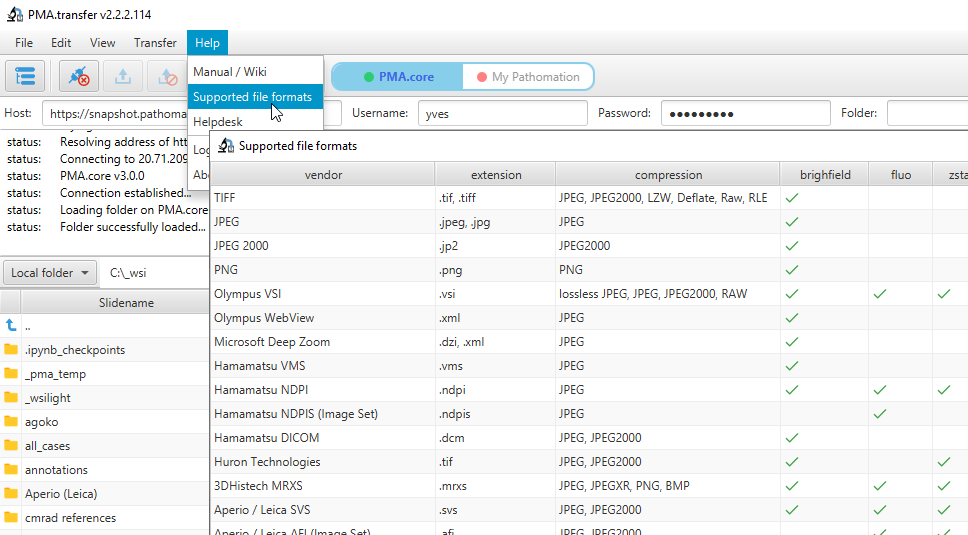
In Python, you can even choose whether to return results as a list, or as a pandas DataFrame object:
Platform meta-data
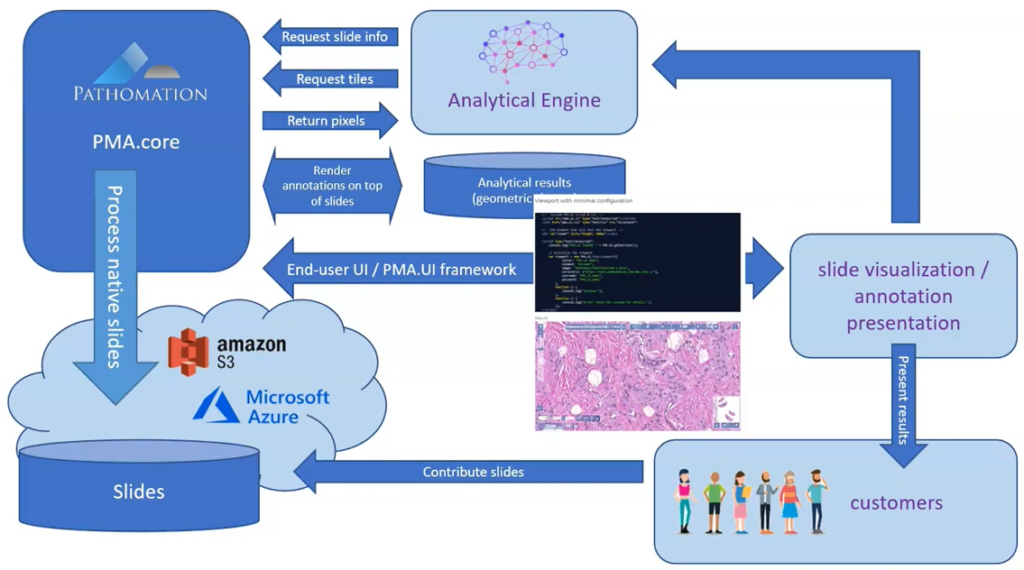
A picture says more than a thousand words, but is worth nothing without being placed in the right context. Without meta-data, those WSI virtual slides stored as gigapixel images are just that: pixels. Pathomation’s software platform for digital pathology (and the PMA.core tile server specifically) offers various opportunities to switch back and forth between (visual) slide metadata and pixels.
This is how Pathomation allows its customers to implement optimal workflows for digital pathology. Interested to switching over to Pathomation so you can exploit this kind of fine-grained granularity and customization too? Contact us today to discuss migration possibilities.