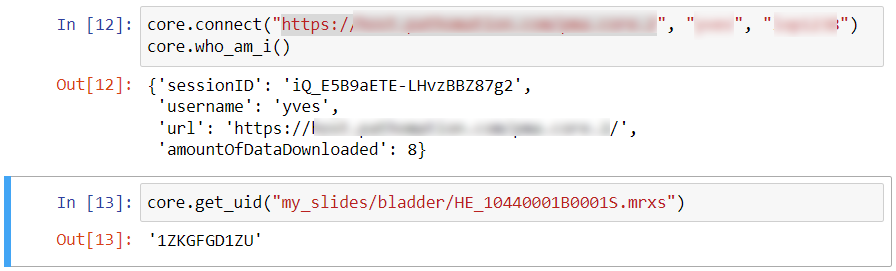
Because sometimes you want more
PMA.studio has a lot of functionality out of the box, but sometimes you want more. Having PMA.studio open on one screen and your organization’s (AP)L(I)MS on another is not always ideal. And what if you don’t have multiple screens?
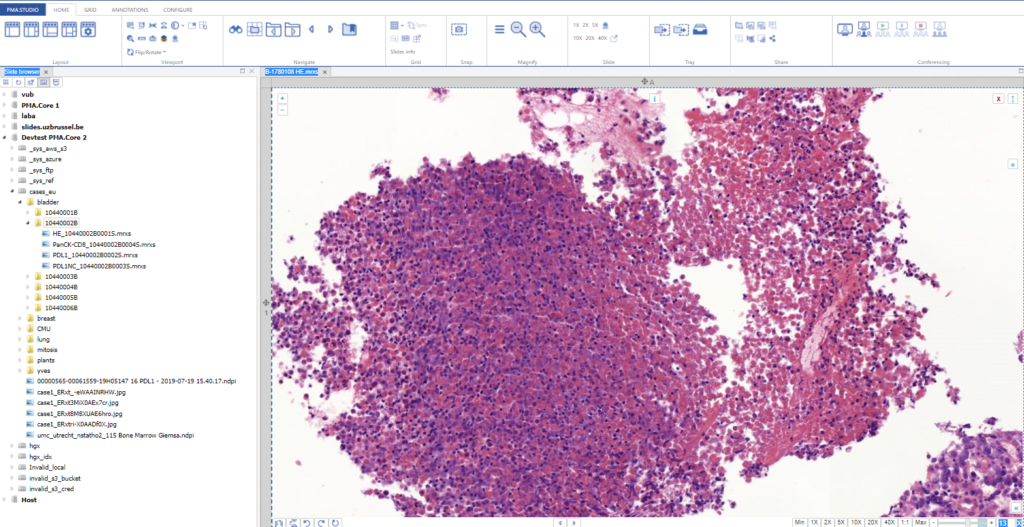
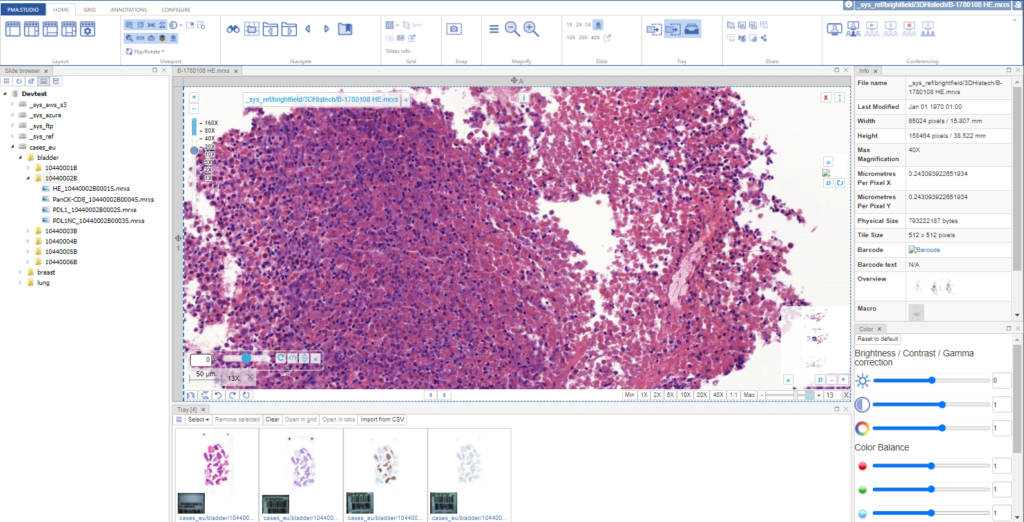
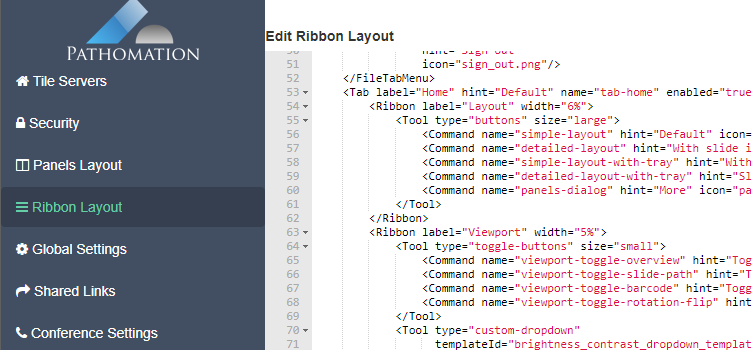
As we pointed out before, PMA.studio offers a panel-based layout. The standard panels can be moved around and even stacked. The standard ribbon in PMA.studio offers some convenient default layouts, too.
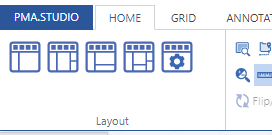
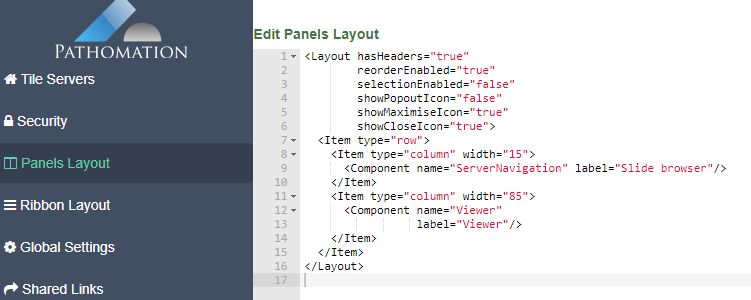
Further configuration of PMA.studio is available through the Configure tab, where you can enable individual panels.
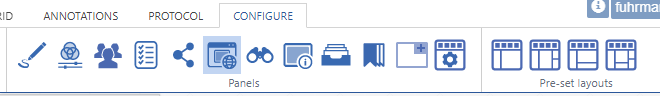
But what if you can’t find the panel that you’re looking for? Maybe the content that you’re looking for is at a different website, and if you just could have that particular page available withing PMA.studio as a separate panel…
Custom panels
PMA.studio offers the possibility the add a custom panel w/ select content from a particular URL.
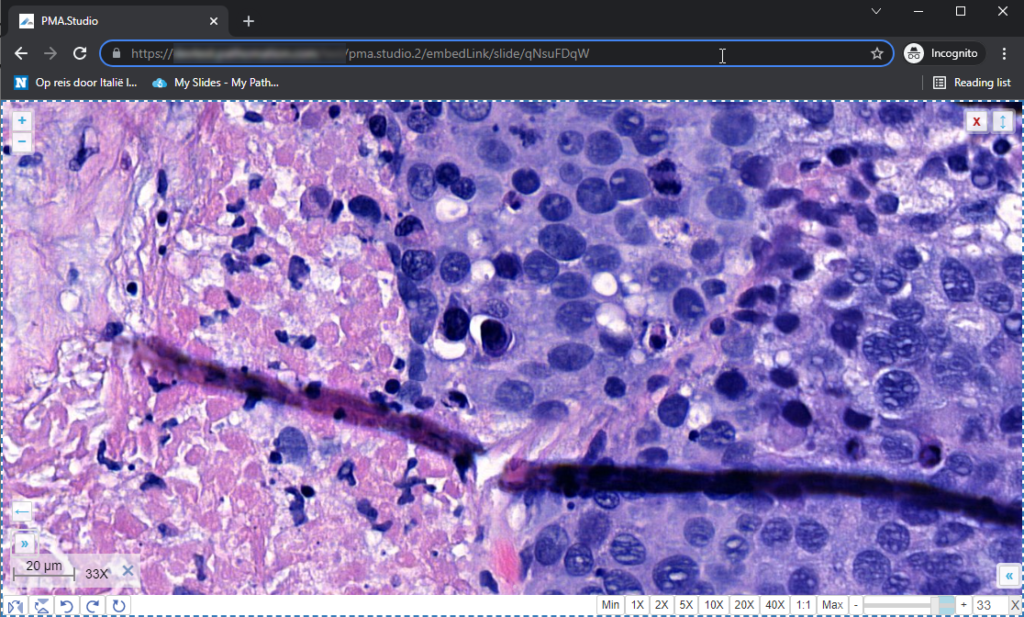
Let’s say that you want to have a reference website available next to your slide content. We’ll use our own PMA.studio wiki website at https://docs.pathomation.com/pma.studio as an example.
You could start by pulling up PMA.studio in one browser, and our wiki in another browser. You start off nice and smooth like this:
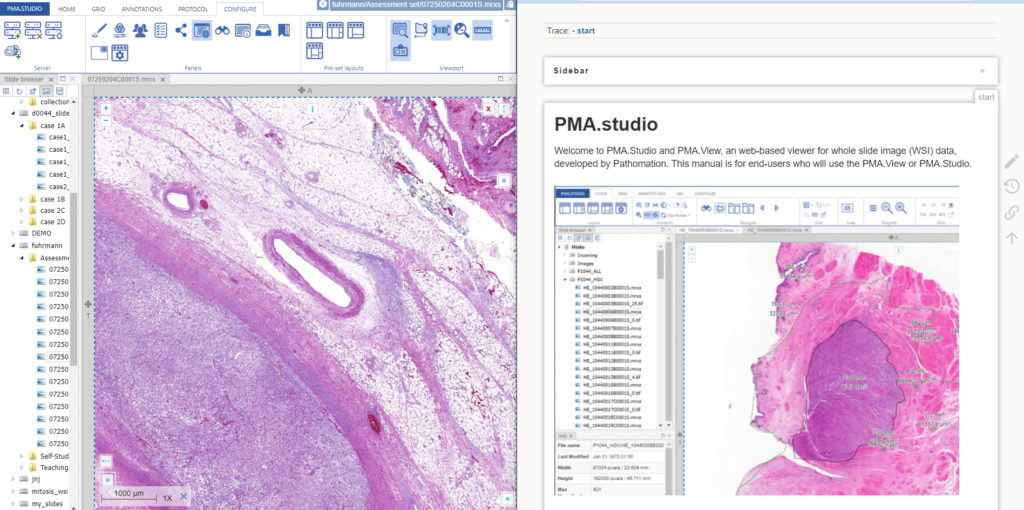
But soon your layout gets messy. The 50% screen width really gives too much to the wiki, and let’s not even get into how easy it is to have that other browser window snowed under a ton of other applications (the word processor you’re using to write a paper in, your ELN, your EPR, your LIMS…)!
There’s a straightforward solution you. Click on the “More” button in the Layout group from the Home tab:

And a new dialog shows up. In addition to selecting any number of default panels, you can also define custom panels. Like so:
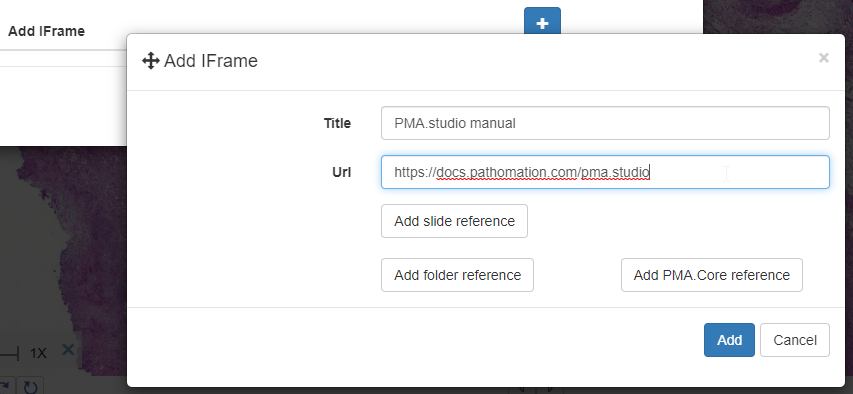
After clicking ok, your new panel appears, making your overall screen layout look like this:
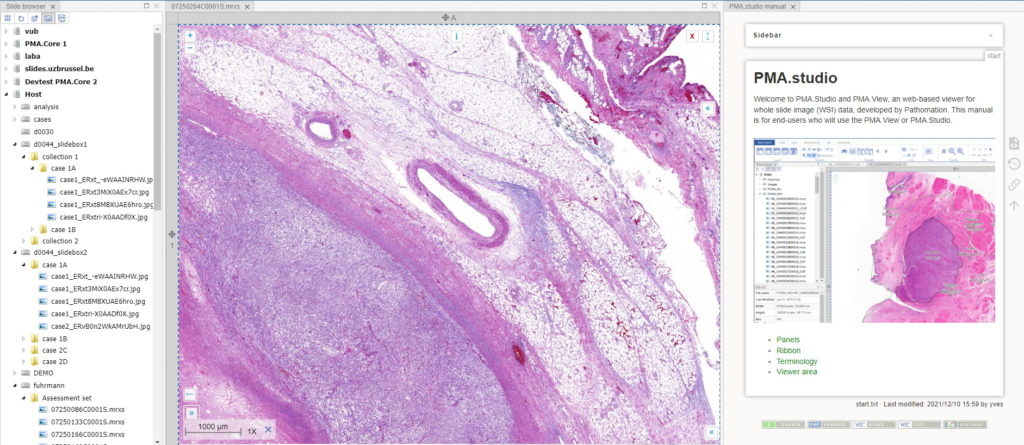
Now that’s more like it!
This is already great for reference data, but what if you want to combine this with slide awareness? In other words: you want to have the content in the panel change automatically depending on the selected slide.
Passing parameters
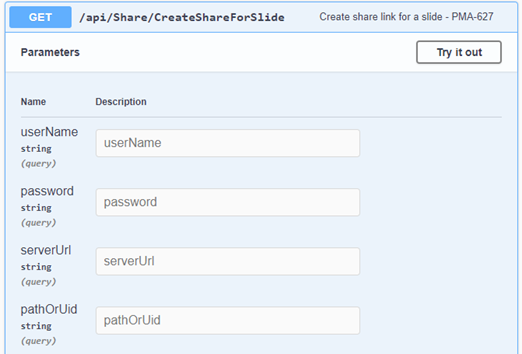
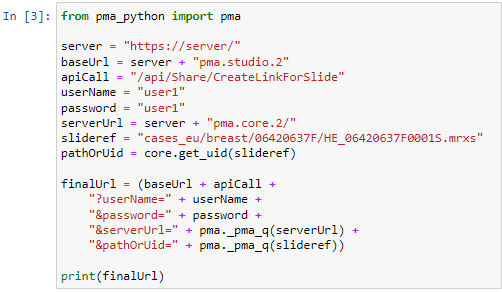
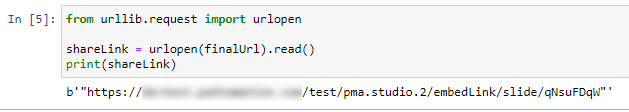
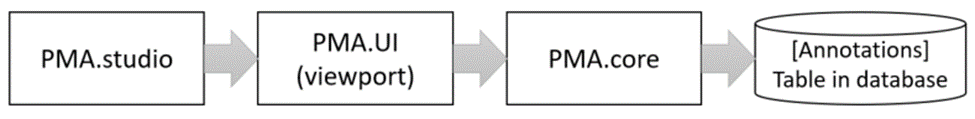
The custom panel mechanism in PMA.studio automatically passes along references to other webpages that trace back to a current PMA.core tile server and selected slide.
This mechanism is typically hidden from plain view to reduce the functional complexity of it all, but a single line of PHP brings up the necessary data:
print_r($_GET);
When we create yet another custom panel that refers to this new page, we see the following appear:
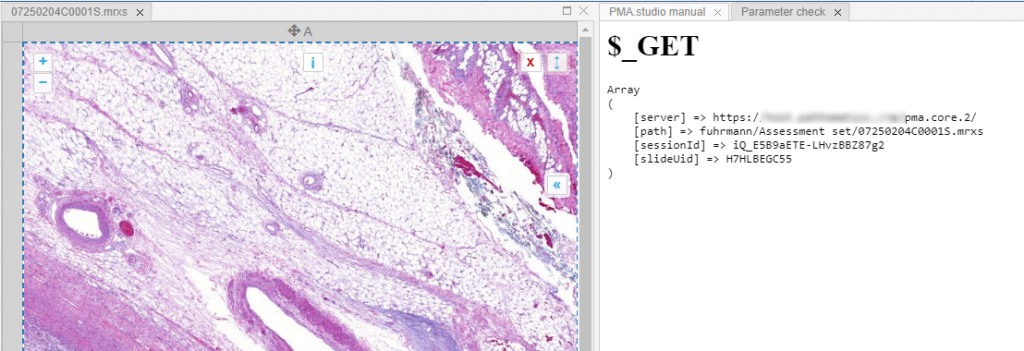
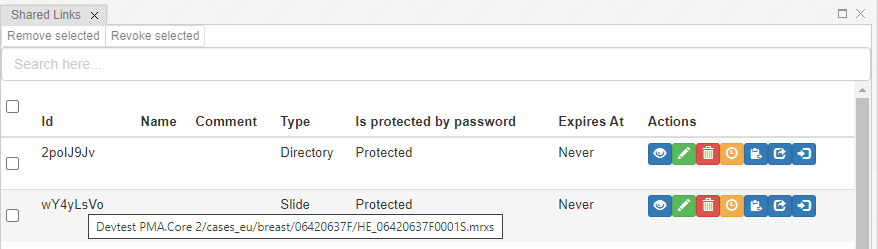
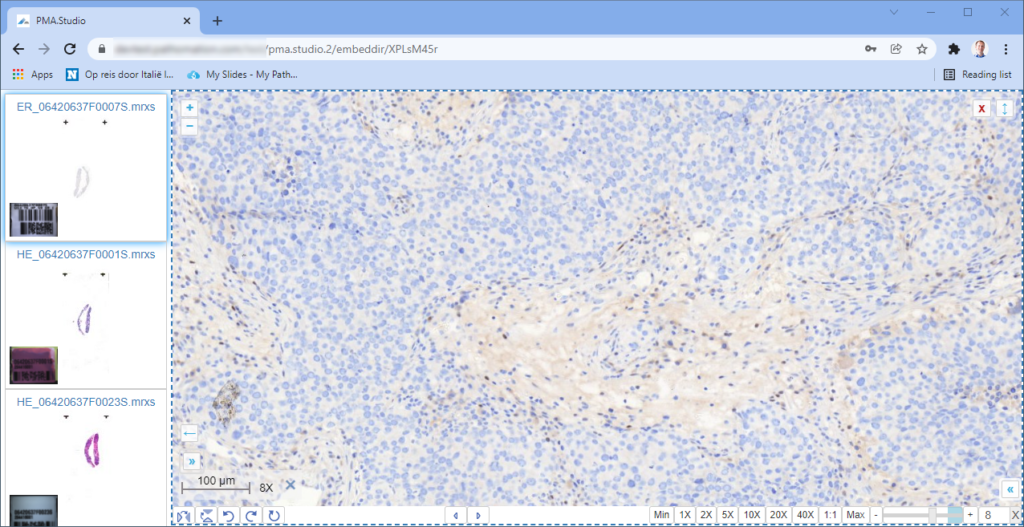
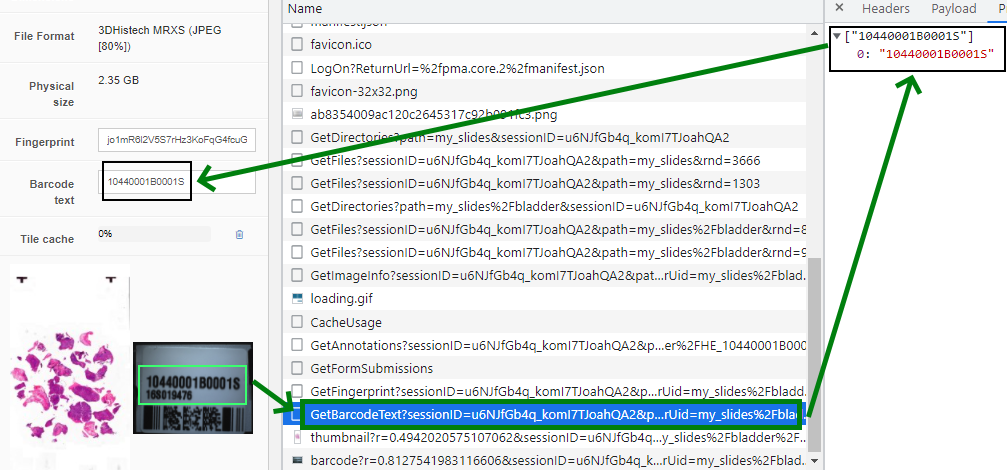
And let’s just say that we don’t like the way PMA.studio displays a slide’s thumbnail and label image. We’d rather just have those in separate panel, too, so we have more control over how they’re displayed.
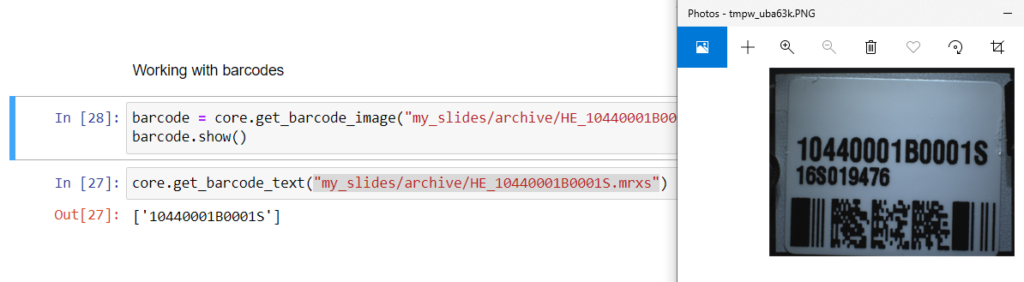
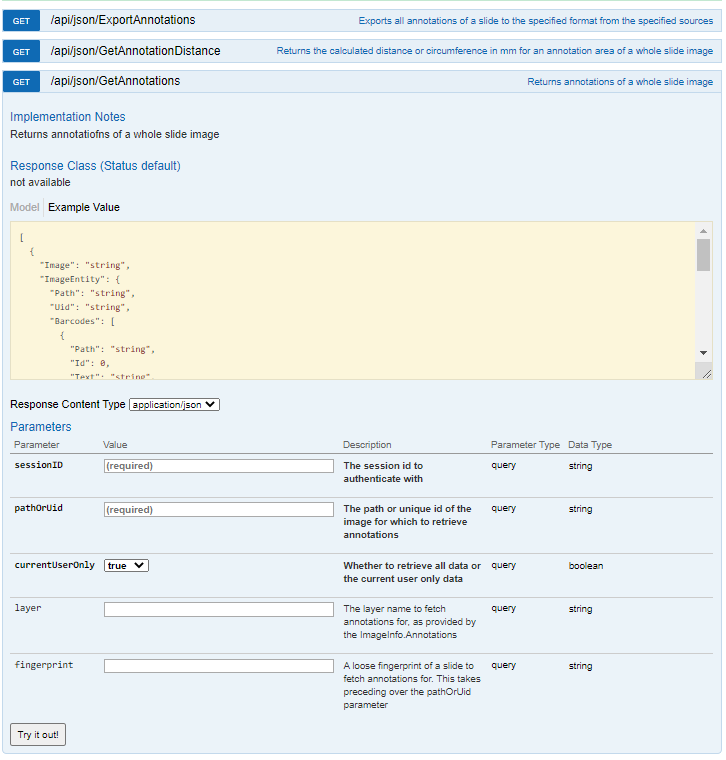
We also know that the thumbnail of slide X can be reached via:
https://server/pma.core/thumbnail?sessionID=…&pathOrUid=X…
And the label image of slide X can be reached via:
https://server/pma.core/barcode?sessionID=…&pathOrUid=X…
We can therefore make two new scripts, that translate the received input parameters from PMA.studio, and translates those to the correct querystring variables for our respective thumbnail and label images:
thumbnail.php:
<?php
$url = $_GET["server"].
"thumbnail?sessionID=".$_GET["sessionId"].
"&pathOrUid=".$_GET["slideUid"];
header("location: $url");
?>
barcode.php:
<?php
$url = $_GET["server"].
"barcode?rotation=0&sessionID=".$_GET["sessionId"].
"&pathOrUid=".$_GET["slideUid"];
header("location: $url");
?>
We place the new files on a server, and reference them via two separate custom panels:
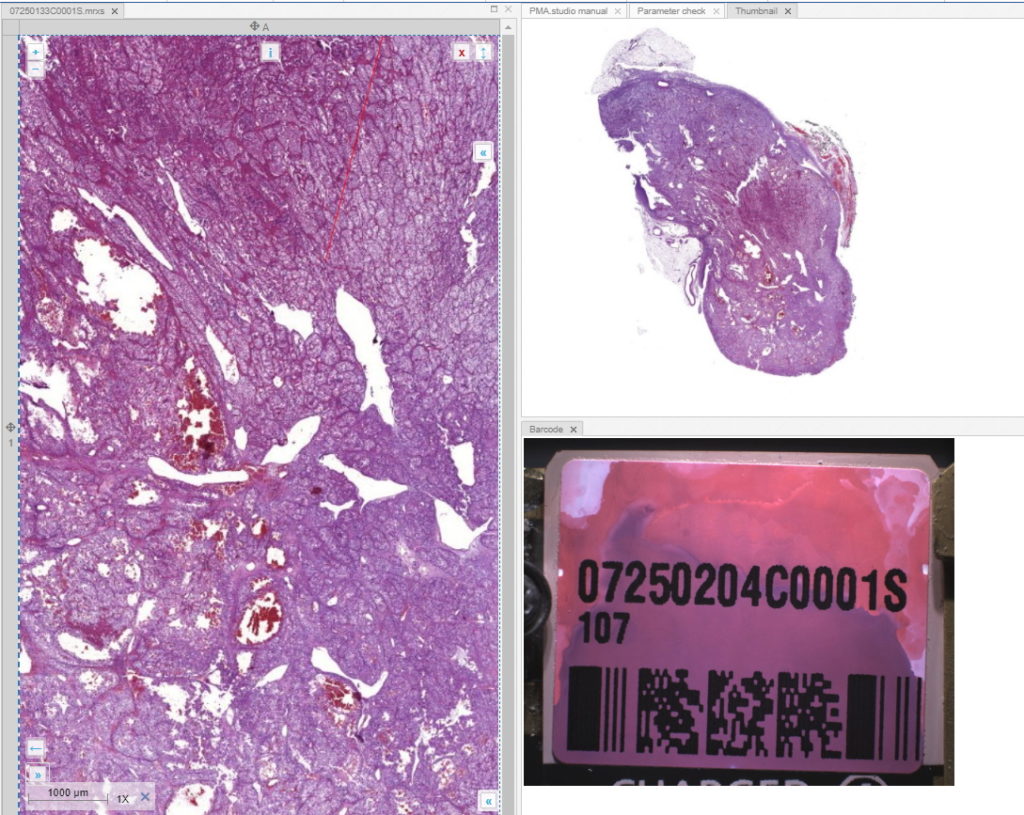
When we navigate the slides in a folder one by one, we now see that the panels are updated accordingly, too.
Other applications
In this post, we showed you how you can configure custom panels in PMA.studio exactly to your liking, and how the content of such panels can synchronize with currently viewed slides.
We showed you how to pass along information through some trivial examples, referring back to our own infrastructure.
Now you can build your interfaces, like we demonstrated in an earlier blog article.
PMA.studio is more than just a universal slide viewer; you can turn it into your own veritable organizational cockpit. Think e.g. about custom database queries against your back-end LIMS, bio-repository, or data warehouse. You can show the data where you want it, when you want it, all with a few configuration tweaks. No longer do you have to juggle multiple browsers, as PMA.studio simply allows you to build your own custom dashboards.
Find out more about PMA.studio through our landing page at https://www.pathomation.com/pma.studio.